
Research
Acquired resistance to targeted therapies in cancer is a rising unmet clinical need. Therapeutic agents exert natural selection and promote the acquisition of new tumor traits. Cellular plasticity endows cancer cells with the ability to overcome therapy by switching their cellular state. At the same time, metabolic dependencies arise in response to alterations in oncogenic growth signaling and might constitute an Achilles heel to be exploited therapeutically. Therapy resistance is a major clinical problem in the treatment of prostate cancer (PCa), in which although androgen deprivation therapy (ADT) has proven effective for its early management, tumors become resistant to ADT, leading to a lethal outcome known as castration-resistant prostate cancer (CRPC). Recent evidence suggests that PCa cells undergo lineage plasticity under therapy pressure to transition from CRPC adenocarcinoma to neuroendocrine prostate cancer (NEPC). This lineage plasticity program is a progressive state in CRPC that involves a switch in cell identity from a luminal to basal phenotype, with low AR signaling and acquired expression of neuroendocrine, neuronal, developmental, and stem cell markers. It has been suggested that reversion of lineage plasticity could restore sensitivity to ADT therapies providing a potential clinical benefit. However, the mechanisms underlying lineage plasticity during NEPC differentiation are not fully understood, and whether this transition state could be targeted therapeutically still needs to be investigated.
Figure 1 Activated stroma in Prostate Cancer
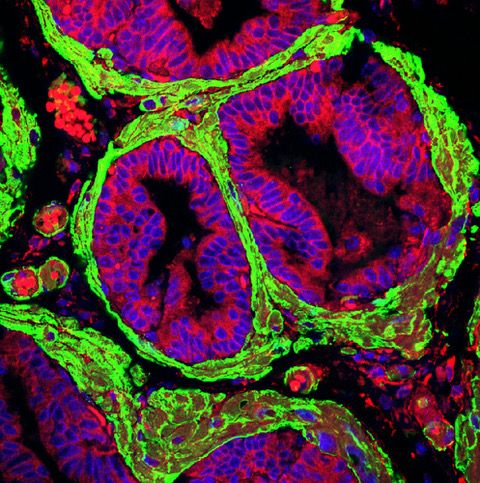
Our laboratory is interested in studying the mechanisms of lineage plasticity during therapy resistance in the context of PKCl/i deficiency. Our published data demonstrate that the loss of the atypical protein kinase C, PKCl/i, results in the metabolic reprogramming of PCa cells to sustain their increased proliferation and epigenetic needs, thus favoring cancer cell plasticity and NEPC differentiation. Our goal is to investigate how kinases orchestrate signaling to link metabolism and epigenetics during the switch in cell identity caused by therapy resistance. In addition, a pending critical question is the contribution of the tumor microenvironment, especially the tumor stroma and the extracellular matrix, to lineage plasticity. Since the loss of PKCl/ialso unleashes a reactive desmoplastic response, we investigate how this kinase is central to integrating cancer cell-intrinsic mechanisms with environmental cues that lead to a plastic state during therapy resistance.
Figure 2 A mouse Prostate Tumor

Current Projects:
- Neuroendocrine Prostate Cancer
- Epigenetic reprogramming
- Tumor microenvironment
- Therapy resistance
- Lineage plasticity
Bio
Maria T. Diaz-Meco earned her Ph.D. in Biochemistry & Molecular Biology at the University Complutense in Madrid. She was an Associate Professor in the National Research Council when she was recruited in 2006 to the Genome Research Institute at the University of Cincinnati. In 2010, she was appointed Professor of Cancer and Cell Biology at the University of Cincinnati Medical College, and in 2011 she joined the Sanford Burnham Prebys Medical Discovery Institute as Professor in the Cancer Metabolism and Signaling Networks Program. Since January 2021, she is a Homer T. Hirst III Professor of Oncology in Pathology at WCM.
Distinctions:
- American Cancer Society Award, Collaboration of the Year (2019)
- Homer T. Hirst III Professor of Oncology (2021)
- Chair of Tumor Host Interactions Study Section, NCI/NIH (2022)