The general workflow of any experiment based on high-throughput DNA sequencing involves the following steps, whose optimal execution requires molecular biology as well as computational expertise.
- Sample prepration This step is usually done by the molecular biologist. What type of starting material is needed depends on the type of assay. For RNA-seq, this would include RNA extraction from all samples of interest; for eRRBS, WGBS, exome-sequencing etc. DNA will have to be extracted; for ChIP-seq, chromatin will be purified, immunoprecipitated and eventually fragmented into small DNA pieces; and so on.
- Sequencing
- Library preparation: the RNA or DNA fragments delivered to the sequencing facility are (highly) amplified, and ligated to the adapters and primers that are needed for sequencing
- Sequencing-by-synthesis: the libraries are loaded onto the lanes of a flow cell, in which the base pair order of every DNA fragment is determined using distinct fluorescent dyes for every nucleotide (for more details, see Section 1.3 of the Introduction to RNA-seq )
- Bioinformatics
- Quality control and processing of the raw sequencing reads, e.g., trimming of excess adapter sequences
- Read alignment and QC
- Additional processing, e.g. normalization to account for differences in sequencing depth (= total numbers of reads) per sample
- Downstream analyses, e.g. identification of differentially expressed genes (RNA-seq), peaks (ChIP-seq), differentially methylated regions (eRRBS, WGBS), sequence variants (exome-seq), and so on
Parameters to consider for the experiment
Here are some of the most important things to think about:
- appropriate control samples (e.g. input samples for ChIP-seq)
- number of replicates
- sequencing read length
- paired-end sequencing or single reads
- specific library preparations, e.g. poly-A enrichment vs. ribosomal depletion for RNA-seq, size range of the fragments to be amplified etc.
- strand information is typically lost, but can be preserved it needed
Typical problems of Illumina-based sequencing data are:
- PCR artifacts such as duplicated fragments, lack of fragments with very high or very low GC content and biases towards shorter read lengths
- sequencing errors and mis-identified bases
These problems can be mitigated, but not completely eliminated, with careful library preparation (e.g., minimum numbers of PCR cycles, removal of excess primers) and frequent updates of Illumina's machines and chemistry.
In addition, there are inherent limitations that are still not overcome:
- short reads -- regions with lots of repetitive regions will be virtually impossible to see with typical read lengths of 100 bp
- the data will most likely be only as good as the reference genome
- statistical analysis of many applications has not caught up with the speed at which new assays are being developed -- gold standards of analysis exist for very few applications and many analyses are still hotly debated in the bioinformatics community (e.g., identification of broad histone mark enrichments, single cell RNA-seq analysis, isoform quantification, de novo transcript discovery (including lncRNA), ...)
There may be questions that are not best addressed with Illumina's sequencing platform!
How many and what types of replicates?
For many HTS applications, the ultimate goal is to find the subset of regions or genes that show differences between the conditions that were analyzed. Conventional RNA-seq analysis, for example, can be used to interrogate thousands of genes at the same time, but the question you're asking for every gene is the same: is there a significant difference in expression between the two (or more) conditions?
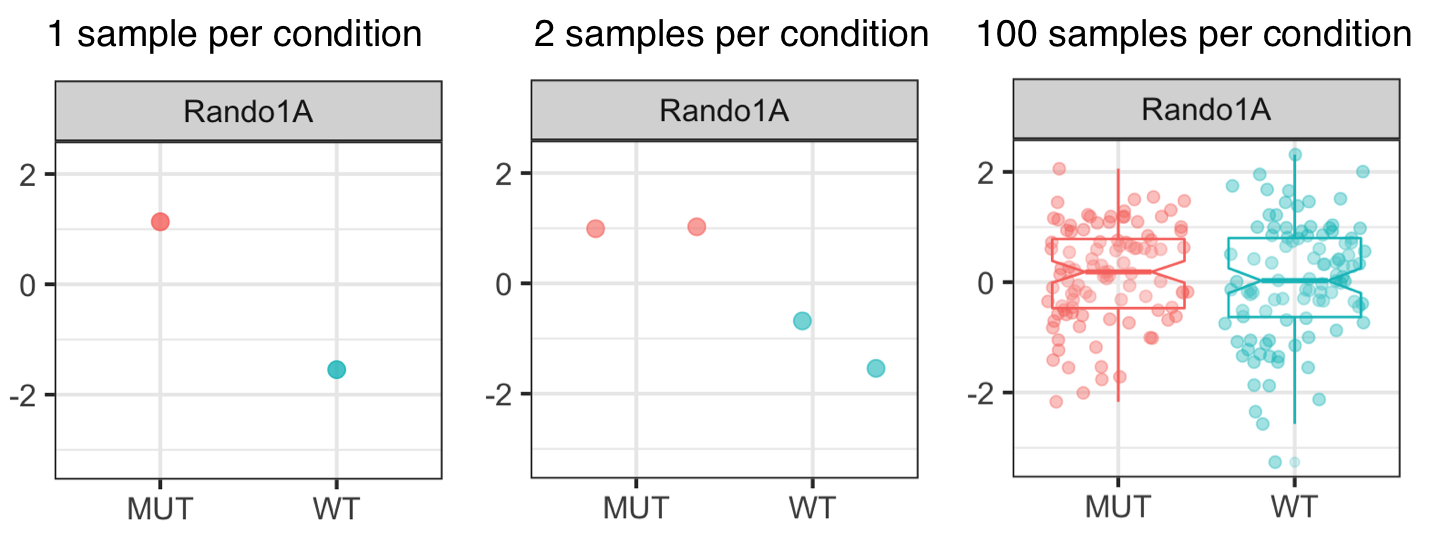
In order to be somewhat confident that the expression levels (or enrichment levels or methylation levels -- or whatever type of biological signal you were interested in) you're comparing are not just reflecting normal biological variation, but are indeed a consequence of the experimental condition, you will need to have more than one measurement per locus. Ideally, you should have hundreds of measurements, but practically, this will not be feasible due to financial and other constraints. You will therefore have to find a compromise between the number of samples you can afford to prepare and sequence and the number of samples you think you will need to gauge the variation in your system. The next figure illustrates how the assessment of a single locus (here called "Rando1A") changes depending on how many and which (!) samples were analyzed (note that all values come from the same distribution that were arbitrarily assigned to either "sample type").
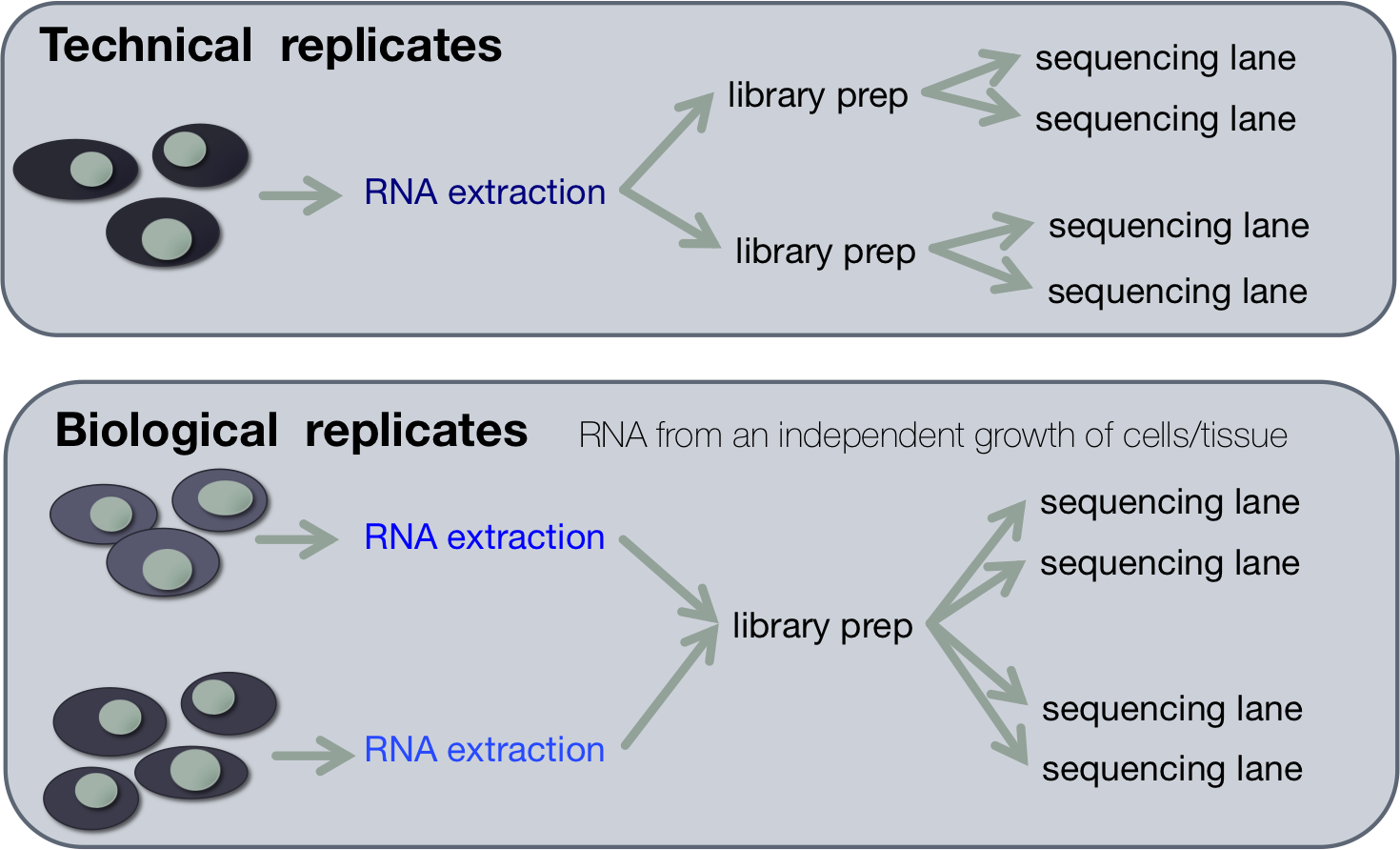
Technical replicates are repeated measurements of the same sample. Biological replicates are parallel measurements of biologically distinct samples that capture random biological variation
Experimental design considerations
The major rule is: Block what you can, randomize what you cannot. In practice, this means that you should try to keep the technical nuisance factors (e.g. cell harvest date, RNA/DNA extraction method, sequencing date, ...) to a minimum, i.e., try to be as consistent as possible. If you cannot harvest all the cells on the same day, make sure you do not confound parameters of interest with technical factors, i.e., absolutely avoid processing all, say, wild type samples on day 1 and all mutant samples on day 2.
Don't overthink it (fully blocked design is simply not feasible), but make sure that the factors of interest are clear. This also means communicating with the sequencing facility about how to randomize technical variation appropriately and in accordance with your experiment's design. The classic paper by Auer & Doerge established the rules of balanced experimental design while leveraging the features of typical high-throughput DNA sequencing platforms. The following figure is taken from their paper:
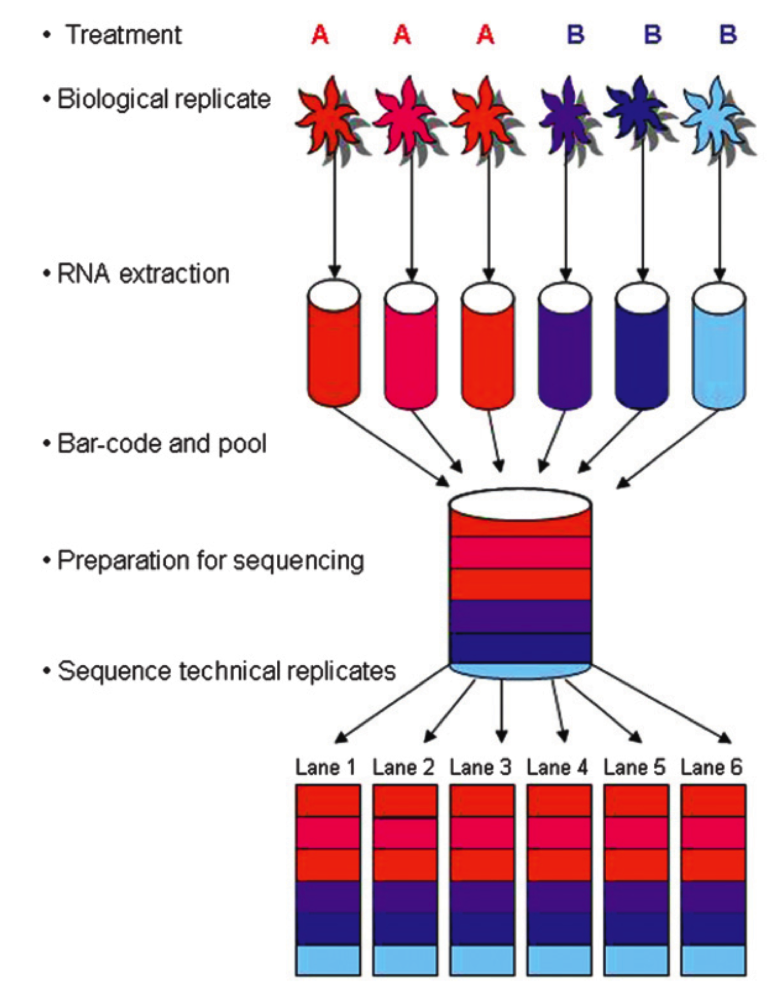
For more details about experimental design considerations, see Section 1.4 of the Introduction to RNA-seq, Altman N and Krzywinski M. (2014) Nature Methods, 12(1):5–6, 2014, and Blainey et al. (2014) Nature Methods, 1(9) 879–880.
For an enlightening read about how even seasoned genomics researchers can fall prey to inadvertend bias, this paper including the comments and discussions is highly recommended.