The human gastrointestinal tract represents an ideal habitat for the growth and proliferation of diverse microbial communities. Next-generation sequencing technologies have provided the opportunity to explore the structure of these communities. Worldwide efforts have delivered solid evidence for the complexity and the cross-kingdom nature of these communities composed of prokaryotes such as bacteria and archaea, eukaryotic viruses and phages, and eukaryotes such as protozoa and fungi.
If you want to know which bacteria are present in your samples (for example, human or mouse fecal samples), two common approaches are 16S Amplicon Sequencing and Shotgun Metagenomics Sequencing. These differ in the type of data you get, as well as in cost per sample. If you want to find out which genes are being actively transcribed by bacteria in your samples, then you should do Metatranscriptomics.
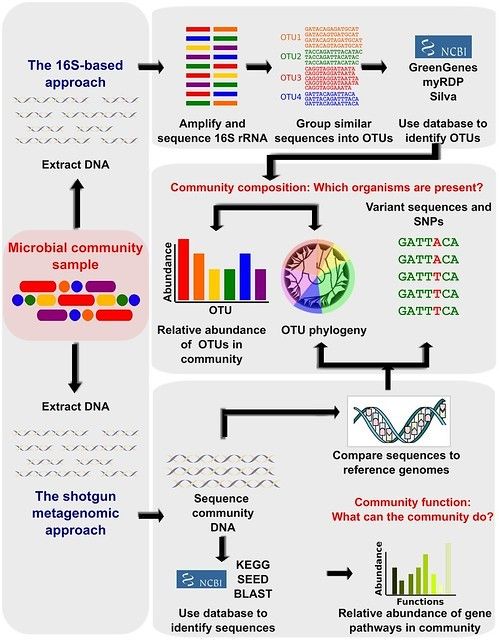
General differences between 16S-based approach, and metagenomics.
By: Morgan and Huttenhower., PLoS Comput Biol 8(12): e1002808.
The most common method to analyze the bacterial composition of samples is 16S Amplicon Sequencing. A specific region common to all bacterial genomes (a variable portion of the 16S ribosomal gene) is amplified and sequenced. After the sequences are processed, a table is provided showing the relative abundance (percentage) of each type of bacteria in each sample. You can then compare groups of samples to see whether their compositions are different. 16S amplicon sequencing is an inexpensive way to get a broad overview of microbiomes. However, the “taxonomic resolution” is limited: in most cases bacteria can not be identified precisely at the species level.
A more detailed picture of a microbiome is provided by Shotgun Metagenomics – untargeted sequencing of all accessible DNA in a sample. The analysis results include a breakdown of the percentage of DNA fragments coming from each type of bacteria – often at a much finer level of classification than is possible with 16S amplicon sequencing. You can also see which genes and metabolic pathways are present. With enough data, more advanced analysis may be possible – for example, whole-genome reconstruction and strain-level analysis. Compared to 16S amplicon sequencing, shotgun metagenomics yields a more complex and detailed picture of the microbial communities in your samples. On the other hand, it is also more expensive and often requires computational expertise to interpret.
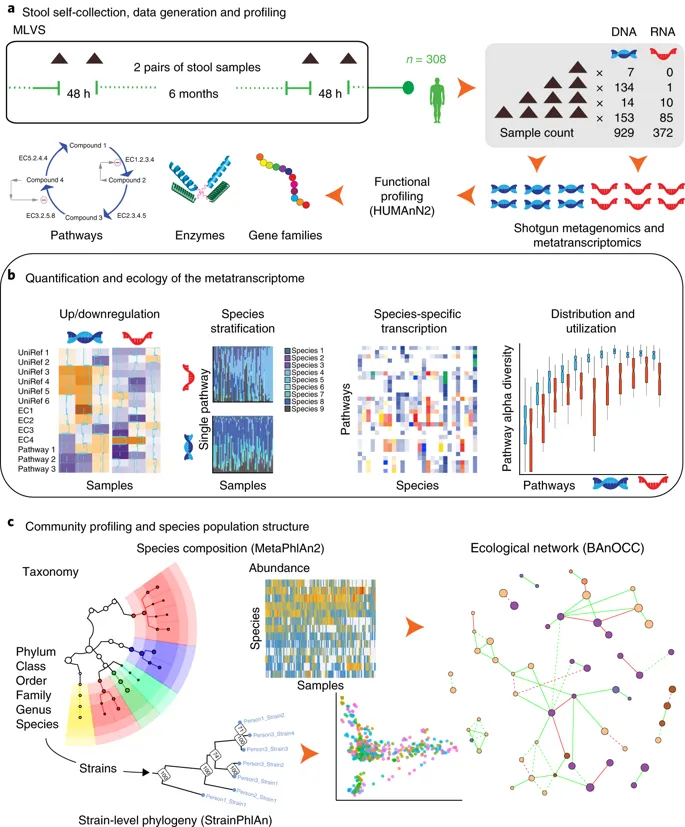
By: Galeb S. Abu-Ali et al., Nature Microbiology, Vol. 3, p356–366 (2018)
While 16S amplicon sequencing and shotgun metagenomics reveal which genomes are present, they say nothing about the bacterial genes being actively transcribed. For that, you need Metatranscriptomics. Here, bacterial messenger RNA is reverse-transcribed, and the resulting DNA fragments are sequenced. Analysis results include abundance tables of bacterial genes and metabolic pathways. This allows you to detect transcriptional changes even when the population of bacteria stays the same.
These services are provided by the Microbiome Core Lab at Weill Cornell Medicine: https://www.microbiome.weill.cornell.edu/
Of course the “microbiome” is complex, harboring not only bacteria, but also viruses (virome), protozoa and fungi (mycobiome), all integrated into a functional trans-kingdom community. The analysis of these fractions requires further enrichment and selection steps, specific methodology, tailored databases, and computational pipelines. Computational integration of all these communities, with their complex functionalities is still in its infancy.
Iliyan D. Iliev, PhD